| World Journal of Nephrology and Urology, ISSN 1927-1239 print, 1927-1247 online, Open Access |
| Article copyright, the authors; Journal compilation copyright, World J Nephrol Urol and Elmer Press Inc |
| Journal website https://wjnu.elmerpub.com |
Case Report
Volume 14, Number 1, June, pages 9-21

TAFRO in Disguise: Idiopathic Multicentric Castleman Disease-Like Syndrome With Renal Thrombotic Microangiopathy and Autoimmune Overlap
Muralidhar Idamakantia, c ![]() , Rani Indrani Bijjama
, Rani Indrani Bijjama ![]() , Thelmo
Fidel Barrantes-Ramirezb
, Thelmo
Fidel Barrantes-Ramirezb
aAdult Inpatient Medical Services (AIMS), Presbyterian Healthcare Services (PHS),
Albuquerque, NM 87106, USA
bRenal Medical Associates, Presbyterian Healthcare
Services (PHS), Albuquerque, NM 87106, USA
cCorresponding Author: Muralidhar
Idamakanti, Adult Inpatient Medical Services (AIMS), Presbyterian Healthcare Services (PHS),
Albuquerque, NM 87106, USA
Manuscript submitted June 1, 2025, accepted June 12, 2025, published online June 16,
2025
Short title: Autoimmune-Driven MCD Complicated by Renal TMA
doi:
https://doi.org/10.14740/wjnu1006
| Abstract | ▴Top |
TAFRO syndrome is a rare systemic inflammatory disorder characterized by thrombocytopenia, anasarca, fever/elevated C-reactive protein (CRP), reticulin fibrosis of bone marrow/renal dysfunction, and organomegaly. It is considered an aggressive subtype of idiopathic multicentric Castleman disease (iMCD), typically requiring lymph node (LN) histopathology for definitive diagnosis. The cases lacking LN biopsy due to accessibility or severe systemic illness but meeting all the criteria for iMCD and TAFRO are categorized as TAFRO with possible iMCD. Another category well recognized in the literature is TAFRO associated with autoimmune diseases, especially Sjogren’s syndrome. We present a 29-year-old female patient with a history of seropositive rheumatoid arthritis, Sjogren’s syndrome, and Barrett’s esophagus who developed an abrupt onset of severe thrombocytopenia, anemia, generalized anasarca, high CRP levels, and renal dysfunction 3 weeks after a viral illness with gastrointestinal symptoms. Imaging revealed hepatosplenomegaly, retroperitoneal lymphadenopathy, and pleural effusion and ascites requiring drainage. Further analysis revealed hypoalbuminemia, elevated soluble interleukin-2 receptor (sIL-2R) and interleukin-6 (IL-6) levels, as well as negative human herpesvirus-8 (HHV-8), cytomegalovirus (CMV), and Epstein-Barr virus (EBV) serologies. ADAM metallopeptidase with thrombospondin type 1 motif 13 (ADAMST13) and complement studies were also negative. A bone marrow biopsy showed evidence of reticulin fibrosis, but an LN biopsy was not feasible. A renal biopsy demonstrated histopathological features consistent with thrombotic microangiopathy (TMA), a finding increasingly recognized in TAFRO. Additionally, elevated anti-Sjogren’s syndrome-related antigen A (SSA) antibodies were found, suggesting a possible autoimmune overlap. Given no LN biopsy, a diagnosis of “TAFRO syndrome with possible iMCD” with autoimmune overlap was entertained. The patient responded well to systemic corticosteroids, plasmapheresis, and anti-interleukin-1 (IL-1) therapy, anakinra. She was discharged in stable condition with improved clinical condition and renal function. The regimen was changed to anti-IL-6 therapy, sarilumab, in the immediate post-discharge period. This case is unique as it fulfills all six criteria of TAFRO syndrome and emphasizes the importance of recognizing it without an LN biopsy, TAFRO syndrome with possible iMCD, particularly when clinical, laboratory, and bone marrow findings support the diagnosis. This case further highlights the diagnostic complexity of TAFRO syndrome and the co-occurrence of renal TMA and autoimmune overlap, suggesting a potential interplay between multiple autoimmune cytokine-driven inflammatory processes. Prompt diagnosis and early immunosuppressive therapy are critical to improve systemic and renal outcomes in such cases with diagnostically ambiguous presentations.
Keywords: TAFRO syndrome; Idiopathic multicentric Castleman disease; Thrombocytopenia; Acute renal failure; Thrombotic microangiopathy; Reticulin fibrosis of bone marrow; Anti-IL-1 therapy; Anakinra; Anti-IL-6 therapy; Tocilizumab; Siltuximab; Cyclosporine
| Introduction | ▴Top |
TAFRO syndrome is a rare systemic inflammatory disorder driven by cytokine storms, primarily interleukin-6 (IL-6), vascular endothelial growth factor (VEGF), tumor necrosis factor-alpha (TNF-α), and interleukin-1β (IL-1β), and is characterized by thrombocytopenia, anasarca, fever or elevated inflammatory markers (C-reactive protein (CRP), erythrocyte sedimentation rate (ESR), ferritin), reticulin fibrosis of the bone marrow (BM) or renal dysfunction, and organomegaly [1-3]. The condition arises from aberrant immune activation, possibly triggered by viral infections, autoimmune dysregulation, or unidentified environmental or genetic factors. TAFRO syndrome is considered a subtype of idiopathic multicentric Castleman disease (iMCD), typically requiring specific lymph node (LN) histopathological characteristics for a definitive diagnosis. However, in some cases, particularly when systemic illness is severe or lymphadenopathy is inaccessible, a biopsy may not be feasible. Cases lacking an LN biopsy but meeting all the criteria for iMCD and TAFRO are classified as “TAFRO with possible iMCD”, and these cases remain diagnostically challenging and complex to treat. Another frequently overlapping category proposed in the literature is “TAFRO associated with autoimmune diseases”, especially Sjogren’s syndrome [3-5]. TAFRO syndrome typically manifests acutely and can lead to multiorgan failure, resulting in poor outcomes if untreated. Early recognition is critical but often delayed due to the overlap with autoimmune diseases, malignancies, infections, and other systemic inflammatory syndromes.
| Case Report | ▴Top |
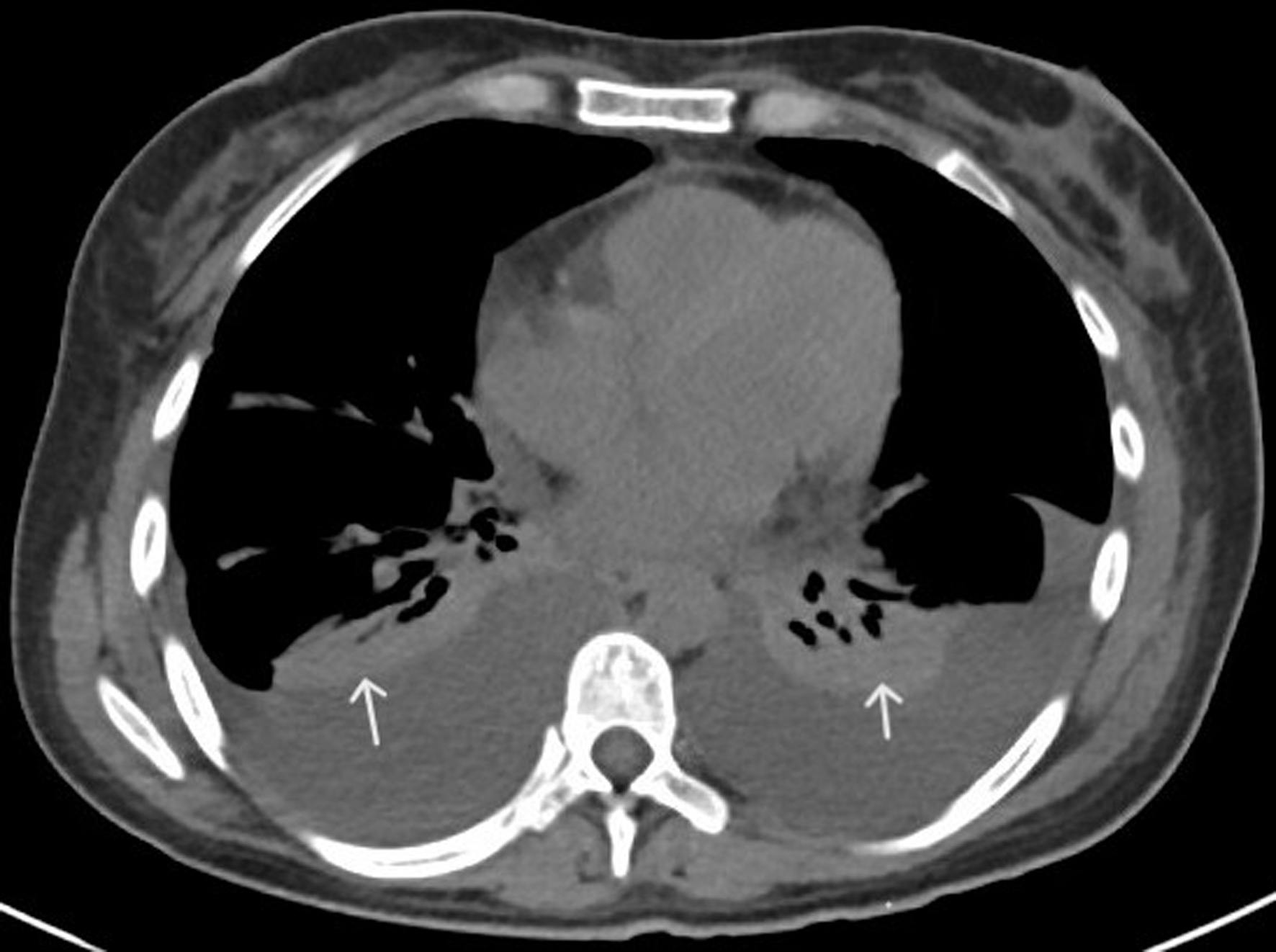
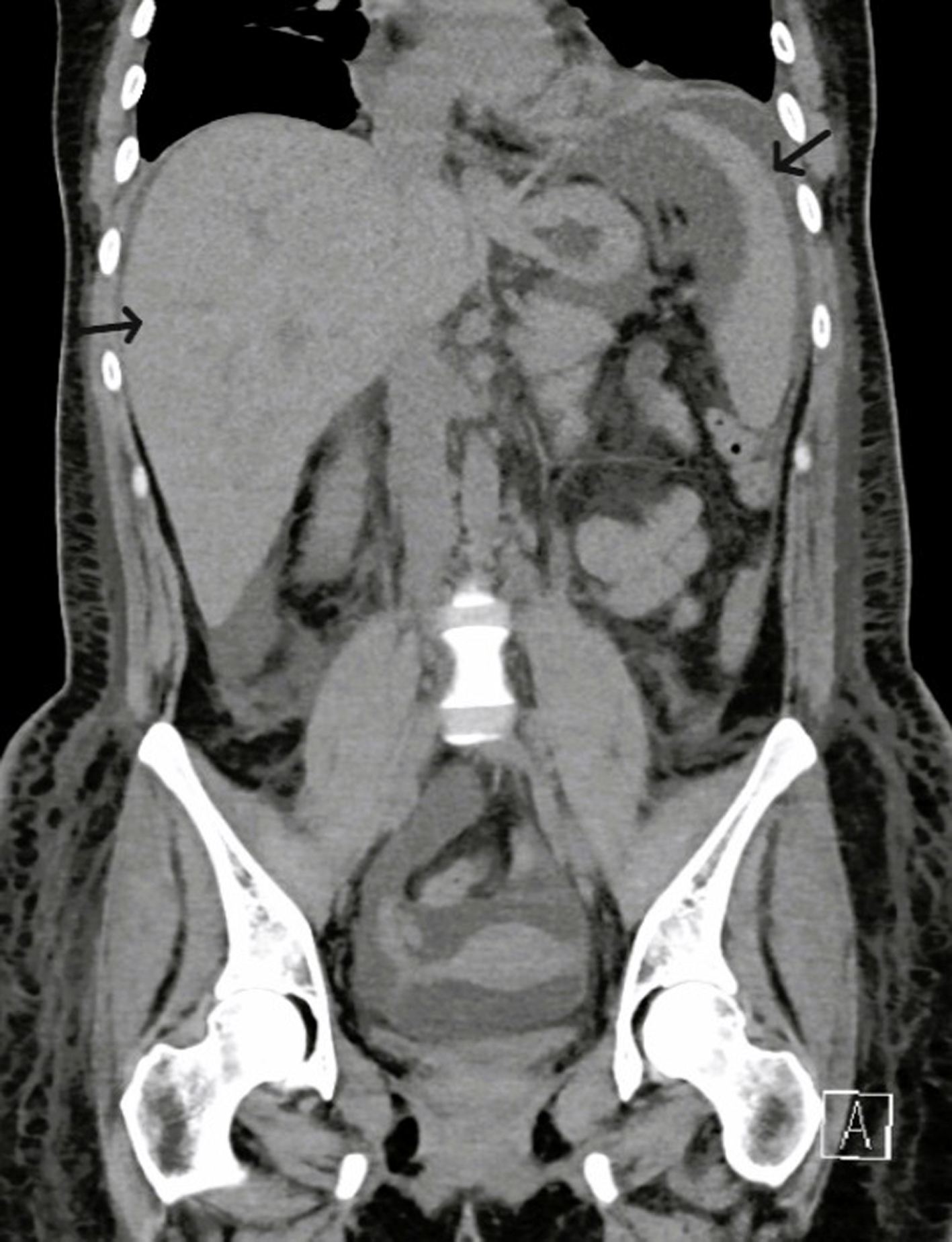
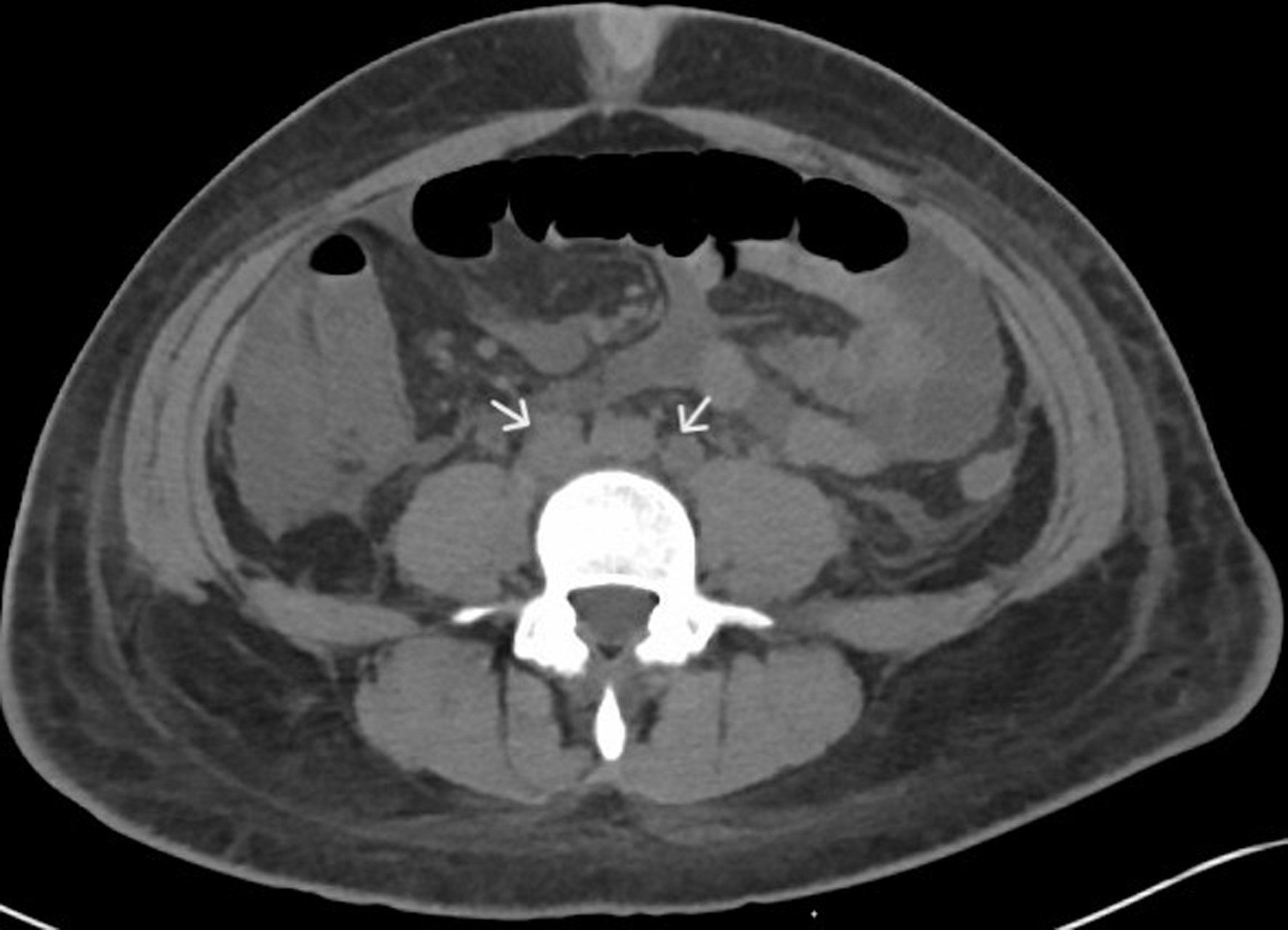
We report a case of a 29-year-old Hispanic-American woman with a history of seropositive rheumatoid arthritis, Sjogren’s syndrome, and Barrett’s disease who presented with the sudden onset of generalized edema, fatigue, decreased urine output, and malaise 2 - 3 weeks after a viral illness with upper respiratory and gastrointestinal symptoms. She appeared acutely ill on initial assessment, showing prominent findings of generalized anasarca (thigh-high lower extremity edema, abdominal distention with abdominal wall edema, decreased breath sounds with crackles, and periorbital facial puffiness), fevers, and generalized pain/myalgias. Initial laboratory results indicated mild leukocytosis (13.9 × 103 cells/µL), mild thrombocytopenia (144 × 103 cells/µL), anemia (9.8 g/dL), hypoalbuminemia (2.2 mg/dL), hyponatremia, and normal liver enzymes (Table 1). A computed tomography (CT) of the chest, abdomen, and pelvis without contrast demonstrated bilateral pleural effusions, ascites, mild hepatosplenomegaly, bilateral perinephric stranding, and small retroperitoneal (diffuse), periaortic, and mesenteric lymphadenopathy (Figs. 1-3).
 Click to view |
Table 1. Laboratory Data on Admission and
Discharge |
 Click for large image |
Figure 1. CT of chest with contrast (axial view) showing bilateral pleural effusions and adjacent atelectasis (white arrows). CT: computed tomography. |
 Click for large image |
Figure 2. CT of abdomen and pelvis with contrast (coronal view) showing mild hepatosplenomegaly (black arrows). CT: computed tomography. |
 Click for large image |
Figure 3. CT of abdomen and pelvis with contrast (axial view) showing retroperitoneal lymphadenopathy (white arrows). CT: computed tomography. |
Urinalysis showed no hematuria and did not indicate a urinary tract infection; however, the patient was empirically initiated on intravenous (IV) ceftriaxone 1 g daily due to the CT findings. Random urine protein was elevated at 157 mg/dL, and the protein-to-creatinine ratio was 0.87. The transthoracic echocardiogram revealed normal ejection fraction and mild pericardial effusion. In addition to diuretics and albumin infusions, the patient underwent both diagnostic and therapeutic thoracentesis and paracentesis, with analysis showing noninfectious reactive fluid and negative cytology. Further laboratory analysis indicated elevated inflammatory markers (CRP, ferritin, ESR) (Table 1). The patient tested negative for influenza A and B, COVID-19, and an extended respiratory panel, including respiratory syncytial virus (RSV). Infectious diseases were consulted, and an infectious mononucleosis-like condition was suspected. The patient had a negative Monospot test and negative serologies for acute hepatitis panel, human herpes virus-8 (HHV-8), human herpes virus-6 (HHV-6), cytomegalovirus (CMV), and Epstein-Barr virus (EBV). The antibiotics were discontinued after an empirical course of 7 days.
The patient experienced worsening thrombocytopenia (50 × 103 cells/µL) and anemia (6.8 g/dL) over the following week. She required a transfusion of one unit of packed red blood cells. Gastroenterology was consulted, and the patient underwent esophago-duodenoscopy and colonoscopy, which revealed an irregular Z-line in the esophagus, mild gastritis, a 4 mm rectal polyp, and internal hemorrhoids; pathology results were negative for Helicobacter pylori, metaplasia, or malignancy. A peripheral blood smear showed low platelet counts with clumping, slightly increased and enlarged megakaryocytes, and elevated polymorphonuclear cells (PMNCs). The hemolytic workup was negative, except for elevated lactate dehydrogenase (LDH). ADAM metallopeptidase with thrombospondin type 1 motif 13 (ADAMST13) and complement levels were also normal. Further analysis indicated elevated cytokine levels (IL-6, IL-8, IL-13, and IL-10) and soluble interleukin-2 receptors (sIL-2Rs). The hematology/oncology team was consulted, and they recommended a BM biopsy based on the overall clinical picture. The BM biopsy revealed increased reticulin fibrosis, megakaryocytic hyperplasia with larger cells, and negative features of hemophagocytic lymphohistiocytosis (HLH) (Fig. 4).
 Click for large image |
Figure 4. Bone marrow biopsy showing increased reticulin fibrosis (black arrows) and megakaryocytic hyperplasia with enlarged cells (blue arrows). |
Around the same time, following the rheumatologist’s recommendation, the patient received 3 days of pulse-dose steroids, IV methylprednisolone 1 g daily, followed by IV methylprednisolone 40 mg twice daily, and was eventually transitioned to prednisone 60 mg daily. A retroperitoneal LN biopsy could not be performed due to inaccessibility. The nephrology team was consulted because the patient also had worsening renal function (creatinine 3.16 mg/dL), and a renal biopsy was recommended based on the evidence of an inflammatory autoimmune pathway. The renal biopsy revealed histopathological features consistent with thrombotic microangiopathy (TMA), a finding increasingly recognized in TAFRO. Additionally, the patient was found to have elevated anti-Sjogren’s syndrome-related antigen A (SSA) antibodies, rheumatoid factor, and anti-cyclic citrullinated protein (CCP) antibodies, and considering her history of Sjogren’s syndrome and rheumatoid arthritis, an autoimmune overlap was suspected to have contributed to the exaggerated autoimmune and inflammatory milieu. Multiple other autoimmune rheumatological assays returned negative results.
The patient fulfilled all clinical and laboratory characteristics of iMCD and met all the major and minor criteria of TAFRO syndrome. However, due to the absence of an LN biopsy, the diagnosis of “TAFRO syndrome with possible iMCD” overlapping with “TAFRO associated with autoimmune disease” was considered, and a plan was made to manage her as iMCD-TAFRO. The patient experienced a prolonged hospital course because of the multidisciplinary evaluations and multimodal management. She received three cycles of plasmapheresis based on the recommendations of the nephrologist and hematologist during her fourth week of stay, spaced over 5 days. Following this, she was started on anti-IL-1 therapy, anakinra 100 mg daily subcutaneously, which she received for 5 days while admitted. Anakinra (IL-1β inhibition) was chosen over IL-6 inhibitors due to the exaggerated cytokine/immune response and co-existing autoimmune conditions.
The patient responded well to systemic corticosteroids, plasmapheresis, and anakinra. She was discharged after 5 weeks of hospitalization in a stable condition, with improved anasarca and renal function. A plan was established for her to continue anakinra for five more days, prednisone at 60 mg daily for three additional weeks, and to have close outpatient follow-ups with rheumatology, nephrology, and hematology. During the follow-up visit 1 week after discharge, she showed complete clinical improvement, with resolution of signs of fluid overload, normal blood counts, and baseline renal function. The rheumatologist changed her regimen to anti-IL-6 therapy, sarilumab at 200 mg subcutaneously every 2 weeks, during the immediate post-discharge period, while continuing prednisone at 60 mg daily. A week after the discharge, Renasight (a comprehensive chronic kidney disease gene panel) was sent. It returned negative, including the complement component 3a receptor 1 (C3AR1) gene involved with atypical hemolytic uremic syndrome (aHUS).
| Discussion | ▴Top |
Castleman disease (CD) refers to a group of rare, heterogeneous lymphoproliferative disorders characterized by LN enlargement and systemic inflammation. It exists in two major forms, unicentric CD (UCD), affecting a single LN region, and multicentric CD (MCD), involving multiple LN areas (Table 2) [1, 4, 5]. When MCD occurs without an identifiable cause (negative HHV-8 infection), it is termed iMCD. iMCD is diagnosed after fulfilling specific clinical, laboratory, and LN histopathological criteria (Table 3) [1, 2, 4-6]. TAFRO syndrome, a subtype of iMCD, is a rare systemic inflammatory disorder characterized by thrombocytopenia, anasarca, fever or elevated inflammatory markers (such as CRP, ESR, ferritin), reticulin fibrosis of the BM or renal dysfunction, and organomegaly [4-9]. TAFRO syndrome typically requires particular LN histopathological characteristics for a definitive diagnosis [7-9]. However, in some cases, especially when systemic illness is severe or lymphadenopathy is inaccessible, a biopsy may not be possible. Cases without an LN biopsy but meeting all the criteria for iMCD and TAFRO are classified as “TAFRO with possible iMCD” [3].
Click to view |
Table 2. Comprehensive Classification of
CD |
Click to view |
Table 3. Diagnostic Criteria for
iMCD |
TAFRO syndrome can present without exhibiting features of iMCD, and in this context, five categories of TAFRO are described in the literature: iMCD-TAFRO, TAFRO with possible iMCD, TAFRO associated with autoimmune diseases, TAFRO associated with infections, and TAFRO-like syndrome [2-4] (Table 4). Unlike classic iMCD, which has a more indolent course, TAFRO syndrome typically manifests acutely and can lead to multiorgan failure if untreated. Early recognition is critical but often delayed due to the overlap with autoimmune diseases, malignancies, infections, and other systemic inflammatory syndromes.
Click to view |
Table 4. Five Types/Categories of TAFRO
Syndrome |
TAFRO syndrome was first reported in 2010 and is believed to be more prevalent in Japan. Due to the rarity of TAFRO syndrome and potential underdiagnosis, especially outside of Japan, comprehensive epidemiological studies are lacking and have a rough incidence estimate of 1 in a million [7]. Increased awareness and reporting are essential to better understand its global prevalence and clinical variations. The precise etiology of TAFRO syndrome remains elusive. Proposed mechanisms include aberrant immune activation, possibly triggered by viral infections (e.g., EBV, CMV), autoimmune dysregulation, or unidentified environmental or genetic factors. Many patients have autoantibodies such as antinuclear antibody (ANA) or anti-SSA, suggesting an autoimmune diathesis [10-12]. Genetic predispositions and polymorphisms in cytokine pathways such as IL-6 and VEGF are under investigation. Upregulation of mechanic target of rapamycin (mTOR) and interferon-1 (IFN-1) responsive genes and Janus kinase-signal transducer and activator of transcription (JAK/STAT) pathways is implicated in some studies, forming the basis for some therapeutic targets [7]. Somatic mitogen-activated extracellular signal-regulated kinase 2 (MEK2) mutation (conferring IL hypersensitivity) and germline runt-related transcription factor 1 (RUNX1) mutation (enhancing hematopoietic self-renewal), both activating the mitogen-activated protein kinase (MAPK) pathway, are found in a study on TAFRO patients, highlighting pathogenetic overlap with histiocytoses and myeloid neoplasms [13].
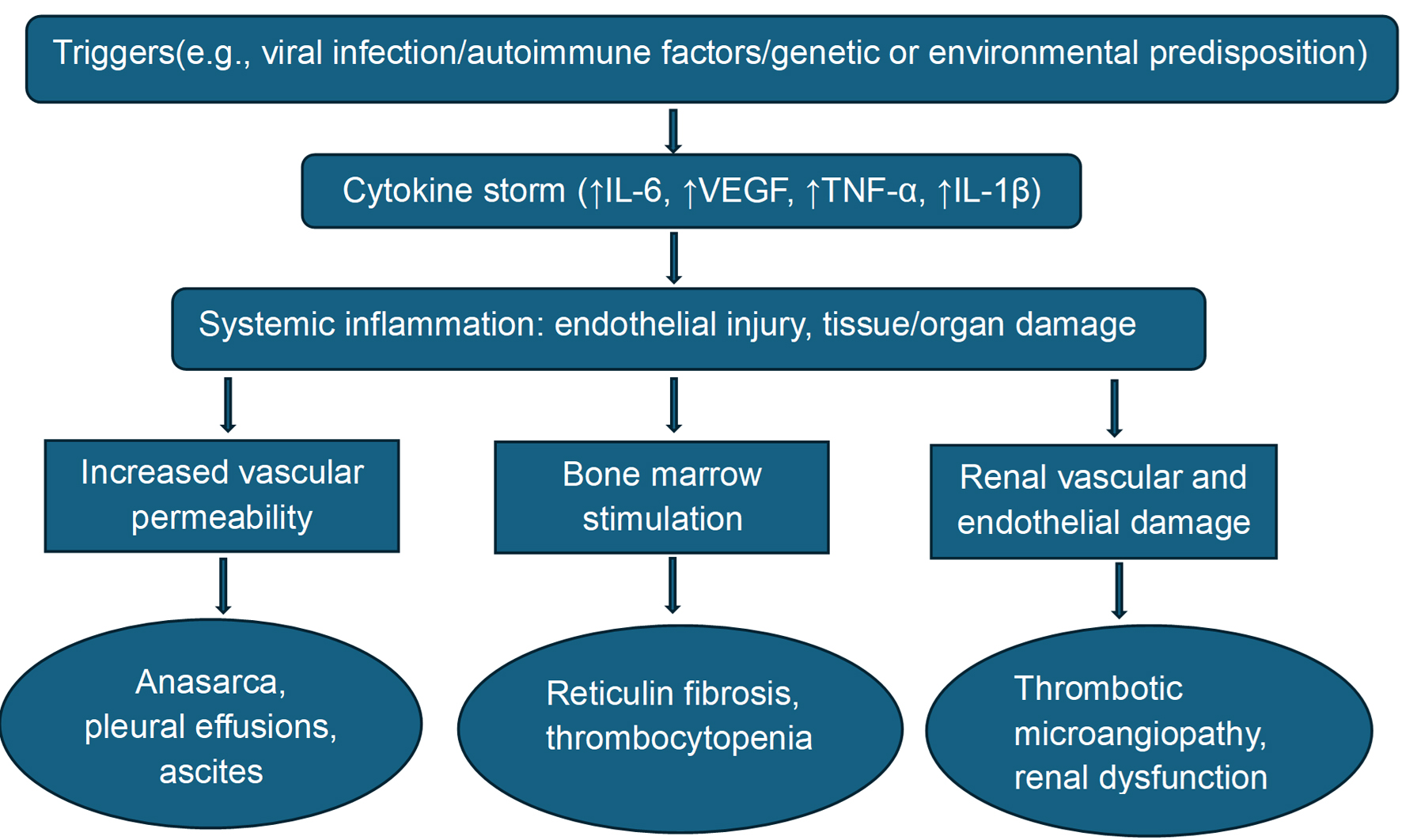
The pathogenesis centers on a hypercytokinemic storm, especially involving IL-6, VEGF, and other proinflammatory mediators such as TNF-α and IL-1β (Fig. 5). IL-6 drives systemic inflammation, anemia, fever, and thrombocytopenia [7, 14, 15]. VEGF increases capillary permeability, leading to anasarca, pleural effusion, and ascites. These cytokines also contribute to glomerular endothelial damage, resulting in renal dysfunction and TMA. BM often reveals reticulin fibrosis and megakaryocytic hyperplasia, signifying a reactive process. Multiple other interleukins (IL-8, IL-10, IL-13) and cell-mediated responses (B-cell, histamines) may also play a role. The interplay of individual susceptibility, cytokine-driven inflammation, endothelial injury, and tissue damage underpins the multiorgan involvement seen in TAFRO [7-9, 14, 15].
 Click for large image |
Figure 5. Pathogenesis of TAFRO syndrome. IL: interleukin; TAFRO: thrombocytopenia, anasarca, fever, reticulin fibrosis/renal failure, and organomegaly; TNF-α: tumor necrosis factor-alpha; VEGF: vascular endothelial growth factor. |
TAFRO typically presents with acute systemic symptoms and evidence of multi-organ involvement, with the initial presentation often being vague and nonspecific. The name TAFRO itself encapsulates most of the associated clinical manifestations: thrombocytopenia, anasarca, effusions, fever, BM suppression, renal dysfunction, and organomegaly [7-9, 14, 15]. BM biopsy shows reticulin fibrosis, and megakaryocytic hyperplasia. Renal biopsy typically shows TMA, and this association with renal failure in TAFRO syndrome is well studied [16-19]. Membranoproliferative glomerulonephritis (MPGN)-like injury is the next common pathological finding found in the biopsy [17, 19]. TAFRO may also present with other nonspecific findings and features of autoimmune disease, as shown in Table 5.
Click to view |
Table 5. Clinical Manifestations of TAFRO
Syndrome |
Diagnosis is based on clinical criteria supplemented by histopathology and exclusion of mimicking diseases. According to the 2019 international criteria, diagnosis of TAFRO is made if the patient meets all three major criteria and at least two of the three minor criteria (Table 6). Our patient in this case report had met all six criteria. Other commonly reported laboratory abnormalities include elevated IL-6, sIL-2R, VEGF, IgA, IgE, LDH, and/or beta-2 microglobulin. Differential diagnoses are broad, and ruling them out is challenging, and forms an essential step towards appropriate diagnosis and timely initiation of management (Table 7) [7, 9, 13, 14].
Click to view |
Table 6. Diagnostic Criteria for TAFRO
Syndrome |
Click to view |
Table 7. Differential Diagnosis of TAFRO
Syndrome |
The management of TAFRO syndrome involves a multimodal immunosuppressive approach tailored to organ dysfunction and disease severity (Tables 8 and 9) [7, 15, 20-46]. There are no major reports or clinical trials that studied the treatment modalities in TAFRO syndrome due to the rarity and sporadic nature of the disease. Various treatment modalities have been tried at various institutions on an individual basis. First-line therapy typically includes high-dose glucocorticoids for rapid control of inflammation [20-22]. In moderate to severe cases, IL-6 inhibitors such as tocilizumab or siltuximab are pivotal in targeting the cytokine-driven pathology for long-term remissions [23-29]. For patients with steroid-refractory disease, cyclosporine may be employed during the acute flares alone or in combination with steroids [30-32]. Rituximab is considered in acute, chronic, and relapse settings, particularly in those with refractory disease, B-cell involvement, or autoimmune overlap [33-37]. Anakinra, an IL-1 receptor antagonist, and canakinumab, a monoclonal antibody against IL-1β, have shown promise in refractory cases, particularly scenarios eliciting exaggerated cytokine storm with autoimmune overlap (Tables 8 and 9) [20, 21, 38-40].
Click to view |
Table 8. Treatment Modalities for TAFRO
Syndrome |
Click to view |
Table 9. Treatment Modalities and Patient
Responses in TAFRO Syndrome |
Chemotherapeutic agents, cyclophosphamide and the CHOP regimen (cyclophosphamide, doxorubicin, vincristine and prednisone), are considered when standard therapies such as corticosteroids, IL-6 inhibitors (e.g., tocilizumab), or cyclosporine fail [41-45, 47]. Use of chemotherapy should be tailored and closely monitored due to the risk of myelosuppression, infections, and organ toxicity. Emerging therapies like JAK inhibitors are still experimental and have been tried in refractory cases [15, 20, 21, 25, 27]. Thalidomide is also considered in sporadic cases unresponsive to commonly tried treatments [41, 48]. Supportive care, including diuretics, albumin infusions, thoracocentesis, paracentesis, renal replacement therapy, electrolyte repletion, antibiotics, plasmapheresis, and transfusions, is critical in managing complications like anasarca, renal dysfunction, electrolyte imbalances, infections, and cytopenias (Tables 8 and 9) [7, 14, 15, 20, 21, 37].
TAFRO syndrome has a variable but often aggressive course, with a subset of patients having a rapidly progressing variant leading to death within a few weeks [49, 50]. Early intervention with targeted immunosuppression has improved outcomes significantly. A proposed prognostic score for iMCD (iMCD-International Prognostic Index (IPI)) could be used to stratify risk in TAFRO patients. In this, patients are divided into three risk groups based on increasing scores on five factors: age > 40, plasmacytic subtype, hepatosplenomegaly, severe anemia (< 8 g/dL), and pleural effusion, with higher scores predicting worse outcomes [50]. Some other studies reported that TAFRO patients with severe renal impairment, persistent thrombocytopenia, or multiorgan failure have a guarded prognosis [20, 21]. Overall survival has been enhanced with IL-6 blockade and multidisciplinary care, but relapses and long-term immunosuppression pose challenges. Five-year survival rates in treated patients range from 65% to 80%, depending on the disease severity and treatment response [20, 21, 49, 51].
Conclusion
This case is unique because it meets all six criteria of TAFRO syndrome and highlights the importance of recognizing TAFRO syndrome with potential iMCD without an LN biopsy, especially when clinical, laboratory, and BM findings support the diagnosis. This case further emphasizes the diagnostic complexity of TAFRO syndrome, which requires a high index of suspicion and acknowledges the co-occurrence of renal TMA and autoimmune overlap syndromes, suggesting a potential interplay among multiple autoimmune cytokine-driven inflammatory processes. First-line treatment options include high-dose glucocorticoids, IL-6 inhibitors, anti-IL-1 drugs, cyclosporine, and rituximab or a combination of these, along with supportive care. Prompt diagnosis and early initiation of immunosuppressive therapy are crucial for improving systemic and renal outcomes in TAFRO syndrome, particularly in cases with diagnostically ambiguous presentations.
Acknowledgments
None to declare.
Financial Disclosure
The authors declare that they do not have a financial relationship with any commercial entity that has an interest in the subject of this manuscript.
Conflict of Interest
The authors declare that they do not have any conflict of interest.
Informed Consent
No patient identifiers or pictures of the patient are used in this manuscript. Verbal consent is obtained from the patient.
Author Contributions
All authors participated actively in this case report. They were actively involved in writing different article sections, helping with images/references, or revising the article before submission. MI (primary) started the article, actively wrote and revised different sections of the article, gathered images, conducted literature review, and added references. RIB identified the rarity of the case report, contributed to different sections of the article, and helped with proofreading (grammar) and revising the article. TFBR actively reviewed, edited, and revised the article, in the capacity of transplant nephrologist.
Data Availability
The authors declare that data supporting the findings of this case report are available within the article.
Abbreviations
ADAMST13: ADAM metallopeptidase with thrombospondin type 1 motif 13; aHUS: atypical hemolytic uremic syndrome; CCP: cyclic citrullinated protein; CD: Castleman disease; CHOP: cyclophosphamide, doxorubicin, vincristine and prednisone; CMV: cytomegalovirus; CRP: C-reactive protein; CT: computed tomography; C3AR1: complement component 3a receptor 1; C3: 4, complement 3, 4; dL: deciliter; ds-DNA: double-stranded deoxyribonucleic acid; EBV: Epstein-Barr virus; ESR: erythrocyte sedimentation rate; HHV-8: human herpesvirus-8; HIV: human immunodeficiency virus; HLH: hemophagocytic lymphohistiocytosis; IFN-1: interferon-1; IL: interleukin; iMCD: idiopathic multicentric Castleman disease; IPI: International Prognostic Index; JAK/STAT: Janus kinase-signal transducer and activator of transcription; LDH: lactate dehydrogenase; LN: lymph node; MAPK: mitogen-activated protein kinase; MAS: macrophage activation syndrome; MCD: multicentric Castleman disease; MEK2: mitogen-activated extracellular signal-regulated kinase 2; mEq: milliequivalent; µL: microliter; mmol: millimole; MPGN: membranoproliferative glomerulonephritis; mTOR: mechanic target of rapamycin; ng: nanogram; pg: picogram; POEMS: polyneuropathy, organomegaly, endocrinopathy, monoclonal gammopathy, and skin changes; RSV: respiratory syncytial virus; RUNX1: runt-related transcription factor 1; sIL-2Rs: soluble interleukin-2 receptors; SSA: Sjogren’s syndrome-related antigen A; TAFRO: thrombocytopenia, anasarca, fever, reticulin fibrosis/renal failure, and organomegaly; TMA: thrombotic microangiopathy; TNF-α: tumor necrosis factor-alpha; UCD: unicentric Castleman disease; VEGF: vascular endothelial growth factor
| References | ▴Top |
- van Rhee F, Stone K, Szmania S, Barlogie B, Singh Z. Castleman
disease in the 21st century: an update on diagnosis, assessment, and therapy. Clin Adv Hematol
Oncol. 2010;8(7):486-498.
pubmed - Fajgenbaum DC, van Rhee F, Nabel CS. HHV-8-negative, idiopathic
multicentric Castleman disease: novel insights into biology, pathogenesis, and therapy. Blood.
2014;123(19):2924-2933.
doi pubmed - Nishimura Y, Fajgenbaum DC, Pierson SK, Iwaki N, Nishikori A, Kawano
M, Nakamura N, et al. Validated international definition of the thrombocytopenia, anasarca,
fever, reticulin fibrosis, renal insufficiency, and organomegaly clinical subtype (TAFRO) of
idiopathic multicentric Castleman disease. Am J Hematol.
2021;96(10):1241-1252.
doi pubmed pmc - Alaggio R, Amador C, Anagnostopoulos I, Attygalle AD, de Oliveira
Araujo IB, Berti E, Bhagat G, et al. Correction: “The 5th edition of The World Health
Organization Classification of Haematolymphoid Tumours: Lymphoid Neoplasms” Leukemia.
2022 Jul;36(7):1720-1748. Leukemia. 2023;37(9):1944-1951.
doi pubmed pmc - van Rhee F, Oksenhendler E, Srkalovic G, Voorhees P, Lim M,
Dispenzieri A, Ide M, et al. International evidence-based consensus diagnostic and treatment
guidelines for unicentric Castleman disease. Blood Adv. 2020;4(23):6039-6050.
doi pubmed pmc - Liu AY, Nabel CS, Finkelman BS, Ruth JR, Kurzrock R, van Rhee F,
Krymskaya VP, et al. Idiopathic multicentric Castleman’s disease: a systematic literature
review. Lancet Haematol. 2016;3(4):e163-e175.
doi pubmed - Kakutani T, Kamada R, Tamai Y. Pathophysiology, treatment, and
prognosis of thrombocytopenia, anasarca, fever, reticulin fibrosis/renal failure, and
organomegaly (TAFRO) syndrome: a review. Curr Issues Mol Biol. 2024;46(10):11255-11269.
doi pubmed pmc - Grange L, Chalayer E, Boutboul D, Paul S, Galicier L, Gramont B,
Killian M. TAFRO syndrome: a severe manifestation of Sjogren’s syndrome? A systematic
review. Autoimmun Rev. 2022;21(8):103137.
doi pubmed - Chen T, Feng C, Zhang X, Zhou J. TAFRO syndrome: a disease that known
is half cured. Hematol Oncol. 2023;41(3):310-322.
doi pubmed - Li ZY, Kim S, Huang S, Mian R. Multicentric Castleman disease with
TAFRO syndrome and Sjogren’s. Clin Case Rep. 2019;7(12):2388-2392.
doi pubmed pmc - Fujimoto S, Kawabata H, Kurose N, Kawanami-Iwao H, Sakai T, Kawanami
T, Fujita Y, et al. Sjogren’s syndrome manifesting as clinicopathological features of
TAFRO syndrome: a case report. Medicine (Baltimore). 2017;96(50):e9220.
doi pubmed pmc - Suzuki E, Ichimura T, Kimura S, Kanno T, Migita K. Primary
Sjogren’s syndrome accompanied by clinical features of TAFRO syndrome. Case Rep
Rheumatol. 2020;2020:8872774.
doi pubmed pmc - Yoshimi A, Trippett TM, Zhang N, Chen X, Penson AV, Arcila ME,
Pichardo J, et al. Genetic basis for iMCD-TAFRO. Oncogene. 2020;39(15):3218-3225.
doi pubmed pmc - Igawa T, Sato Y. TAFRO syndrome. Hematol Oncol Clin North Am.
2018;32(1):107-118.
doi pubmed - Caballero JC, Conejero N, Solan L, Diaz de la Pinta FJ, Cordoba R,
Lopez-Garcia A. Unraveling TAFRO syndrome: an in-depth look at the pathophysiology, management,
and future perspectives. Biomedicines. 2024;12(5):1076.
doi pubmed pmc - Tu KH, Fan PY, Chen TD, Chuang WY, Wu CY, Ku CL, Tian YC, et al.
TAFRO syndrome with renal thrombotic microangiopathy: insights into the molecular mechanism and
treatment opportunities. Int J Mol Sci. 2021;22(12):6286.
doi pubmed pmc - Iwasaki T, Mizusaki K, Masumoto M, Minagawa Y, Azuma K, Furukawa T,
Yoshida M, et al. TAFRO syndrome with renal biopsy successfully treated with steroids and
cyclosporine: a case report. BMC Nephrol. 2022;23(1):262.
doi pubmed pmc - Shah N, Davidson T, Cheung C, Keung K. To and TAFRO - a cryptic cause
of acute renal failure: a case report. BMC Nephrol. 2022;23(1):19.
doi pubmed pmc - Leurs A, Gnemmi V, Lionet A, Renaud L, Gibier JB, Copin MC, Hachulla
E, et al. Renal pathologic findings in TAFRO syndrome: is there a continuum between thrombotic
microangiopathy and membranoproliferative glomerulonephritis? A case report and literature
review. Front Immunol. 2019;10:1489.
doi pubmed pmc - Miura K, Nishimaki-Watanabe H, Takahashi H, Nakagawa M, Otake S,
Hamada T, Koike T, et al. TAFRO syndrome: guidance for managing patients presenting
thrombocytopenia, anasarca, fever, reticulin fibrosis, renal insufficiency, and organomegaly.
Biomedicines. 2024;12(6):1277.
doi pubmed pmc - Pierson SK, Lim MS, Srkalovic G, Brandstadter JD, Sarmiento
Bustamante M, Shyamsundar S, Mango N, et al. Treatment consistent with idiopathic multicentric
Castleman disease guidelines is associated with improved outcomes. Blood Adv.
2023;7(21):6652-6664.
doi pubmed pmc - van Rhee F, Voorhees P, Dispenzieri A, Fossa A, Srkalovic G, Ide M,
Munshi N, et al. International, evidence-based consensus treatment guidelines for idiopathic
multicentric Castleman disease. Blood. 2018;132(20):2115-2124.
doi pubmed pmc - van Rhee F, Wong RS, Munshi N, Rossi JF, Ke XY, Fossa A, Simpson D,
et al. Siltuximab for multicentric Castleman’s disease: a randomised, double-blind,
placebo-controlled trial. Lancet Oncol. 2014;15(9):966-974.
doi pubmed - Marchetti M, Feyles E, Zinzani P. Frontline siltuximab and rituximab
in TAFRO syndrome: a case report. Eur J Haematol. 2020;105(4):505-507.
doi pubmed - Pierson SK, Shenoy S, Oromendia AB, Gorzewski AM, Langan Pai RA,
Nabel CS, Ruth JR, et al. Discovery and validation of a novel subgroup and therapeutic target in
idiopathic multicentric Castleman disease. Blood Adv. 2021;5(17):3445-3456.
doi pubmed pmc - Cordero L, Aguilar-Rodriguez F, Sandino J, Alonso M, Gutierrez E.
Siltuximab monotherapy in TAFRO syndrome: a case report and review of the literature.
J Nephrol. 2023;36(4):1181-1185.
doi pubmed - Lust H, Gong S, Remiker A, Rossoff J. Idiopathic multicentric
Castleman disease with TAFRO clinical subtype responsive to IL-6/JAK inhibition: a pediatric
case series. Pediatr Blood Cancer. 2021;68(10):e29261.
doi pubmed - Akiyama M, Kaneko Y, Takeuchi T. Tocilizumab for the treatment of
TAFRO syndrome: a systematic literature review. Ann Hematol. 2020;99(11):2463-2475.
doi pubmed - Sakai K, Maeda T, Kuriyama A, Shimada N, Notohara K, Ueda Y. TAFRO
syndrome successfully treated with tocilizumab: a case report and systematic review. Mod
Rheumatol. 2018;28(3):564-569.
doi pubmed - Takasawa N, Sekiguchi Y, Takahashi T, Muryoi A, Satoh J, Sasaki T. A
case of TAFRO syndrome, a variant of multicentric Castleman’s disease, successfully
treated with corticosteroid and cyclosporine A. Mod Rheumatol. 2019;29(1):198-202.
doi pubmed - Inoue M, Ankou M, Hua J, Iwaki Y, Hagihara M, Ota Y. Complete
resolution of TAFRO syndrome (thrombocytopenia, anasarca, fever, reticulin fibrosis and
organomegaly) after immunosuppressive therapies using corticosteroids and cyclosporin A: a case
report. J Clin Exp Hematop. 2013;53(1):95-99.
doi pubmed - Yamaga Y, Tokuyama K, Kato T, Yamada R, Murayama M, Ikeda T, Yamakita
N, et al. Successful treatment with cyclosporin A in tocilizumab-resistant TAFRO syndrome.
Intern Med. 2016;55(2):185-190.
doi pubmed - Servati S, Mohammadi I, Rajai Firouzabadi S. First-line treatment of
TAFRO syndrome with rituximab: a case report and literature review. Ann Hematol.
2025;104(5):3035-3046.
doi pubmed pmc - Mian H, Leber B. Mixed variant multicentric Castleman disease treated
with rituximab: case report. J Pediatr Hematol Oncol. 2010;32(8):622.
doi pubmed - Jain P, Verstovsek S, Loghavi S, Jorgensen JL, Patel KP, Estrov Z,
Fayad L, et al. Durable remission with rituximab in a patient with an unusual variant of
Castleman’s disease with myelofibrosis-TAFRO syndrome. Am J Hematol.
2015;90(11):1091-1092.
doi pubmed pmc - Tsurumi H, Fujigaki Y, Yamamoto T, Iino R, Taniguchi K, Nagura M,
Arai S, et al. Remission of refractory ascites and discontinuation of hemodialysis after
additional rituximab to long-term glucocorticoid therapy in a patient with TAFRO syndrome.
Intern Med. 2018;57(10):1433-1438.
doi pubmed pmc - Sakaki A, Hosoi H, Kosako H, Furuya Y, Iwamoto R, Hiroi T, Murata S,
et al. Successful combination treatment with rituximab, steroid pulse therapy, plasma exchange
and romiplostim for very severe TAFRO syndrome. Leuk Lymphoma. 2022;63(10):2499-2502.
doi pubmed - Galeotti C, Tran TA, Franchi-Abella S, Fabre M, Pariente D, Kone-Paut
I. IL-1RA agonist (anakinra) in the treatment of multifocal Castleman disease: case report.
J Pediatr Hematol Oncol. 2008;30(12):920-924.
doi pubmed - Lanzillotta M, Sant'Angelo M, Kaneko N, Pillai S, Ponzoni M,
Della-Torre E. Treating life-threatening TAFRO syndrome with interleukin-1 inhibition.
Eur J Intern Med. 2021;87:121-123.
doi pubmed - Palmeri S, Ferro J, Natoli V, Matucci-Cerinic C, Papa R, Rosina S,
Sorrentino S, et al. Efficacy of high-dose intravenous anakinra in pediatric TAFRO syndrome:
report of two cases and literature review. Pediatr Blood Cancer. 2025.
doi pubmed - Zhang L, Zhao AL, Duan MH, Li ZY, Cao XX, Feng J, Zhou DB, et al.
Phase 2 study using oral thalidomide-cyclophosphamide-prednisone for idiopathic multicentric
Castleman disease. Blood. 2019;133(16):1720-1728.
doi pubmed - Zhao H, Zhang MY, Shen KN, Feng J, Cao XX, Duan MH, Zhou DB, et al. A
phase 2 prospective study of bortezomib, cyclophosphamide, and dexamethasone in patients with
newly diagnosed iMCD. Blood. 2023;141(21):2654-2657.
doi pubmed - Zhou B, Tang C, Chen G, Jiang T, Shi X, Wang F. TAFRO syndrome with
fatigue and abdominal distension as the first symptom: a case report. Hematol Oncol.
2023;41(2):285-290.
doi pubmed - Zhang Y, Suo SS, Yang HJ, Zhou XP, You LS, Yu WJ, Wang ZM, et al.
Clinical features and treatment of 7 Chinese TAFRO syndromes from 96 de novo Castleman
diseases: a 10-year retrospective study. J Cancer Res Clin Oncol.
2020;146(2):357-365.
doi pubmed pmc - Wu CB, Zhang HY, Shao SH, Dou LW, Zhou QY, Liu Y, Gao WB, et al. [A
report of six TAFRO syndrome: clinical characteristics, diagnosis and treatment analysis].
Zhonghua Yi Xue Za Zhi. 2020;100(8):624-628.
doi pubmed - Morino J, Hirai K, Yoshida K, Kako S, Ookawara S, Oshiro H, Sugawara
H, et al. Successful treatment of TAFRO (thrombocytopenia, anasarca, fever, renal insufficiency,
and organomegaly) syndrome with triple combination therapy of corticosteroid, tocilizumab, and
cyclosporine: a case report. Cureus. 2025;17(3):e80274.
doi pubmed pmc - Yasuda S, Tanaka K, Ichikawa A, Watanabe K, Uchida E, Yamamoto M,
Yamamoto K, et al. Aggressive TAFRO syndrome with reversible cardiomyopathy successfully treated
with combination chemotherapy. Int J Hematol. 2016;104(4):512-518.
doi pubmed - Tatekawa S, Umemura K, Fukuyama R, Kohno A, Taniwaki M, Kuroda J,
Morishita Y. Thalidomide for tocilizumab-resistant ascites with TAFRO syndrome. Clin Case Rep.
2015;3(6):472-478.
doi pubmed pmc - Fajgenbaum DC, Pierson SK, Kanhai K, Bagg A, Alapat D, Lim MS,
Lechowicz MJ, et al. The disease course of Castleman disease patients with fatal outcomes in the
ACCELERATE registry. Br J Haematol. 2022;198(2):307-316.
doi pubmed pmc - Zietz C, Bogner JR, Goebel FD, Lohrs U. An unusual cluster of cases
of Castleman’s disease during highly active antiretroviral therapy for AIDS.
N Engl J Med. 1999;340(24):1923-1924.
doi pubmed - Yu L, Shi M, Cai Q, Strati P, Hagemeister F, Zhai Q, Li L, et al. A
novel predictive model for idiopathic multicentric Castleman disease: The International
Castleman Disease Consortium Study. Oncologist. 2020;25(11):963-973.
doi pubmed pmc
This
article is distributed under the terms of the Creative Commons Attribution Non-Commercial 4.0
International License, which permits unrestricted non-commercial use, distribution, and
reproduction in any medium, provided the original work is properly cited.
World
Journal of Nephrology and Urology is published by Elmer Press Inc.