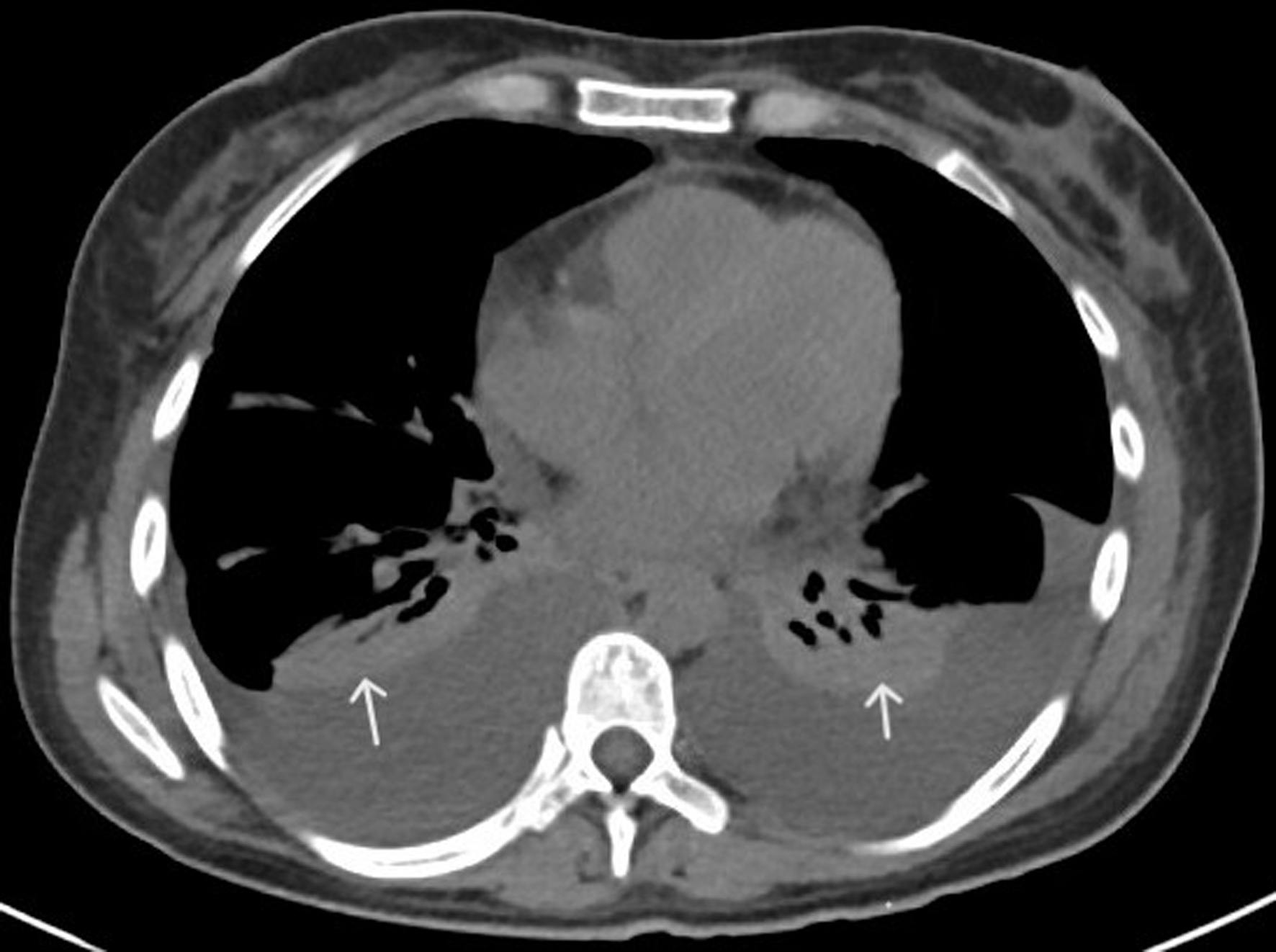
↓ Figure 1. CT of chest with contrast (axial
view) showing bilateral pleural effusions and adjacent atelectasis (white arrows). CT: computed
tomography.
| World Journal of Nephrology and Urology, ISSN 1927-1239 print, 1927-1247 online, Open Access |
| Article copyright, the authors; Journal compilation copyright, World J Nephrol Urol and Elmer Press Inc |
| Journal website https://wjnu.elmerpub.com |
Case Report
Volume 14, Number 1, June, pages 9-21
TAFRO in Disguise: Idiopathic Multicentric Castleman Disease-Like Syndrome With Renal Thrombotic Microangiopathy and Autoimmune Overlap
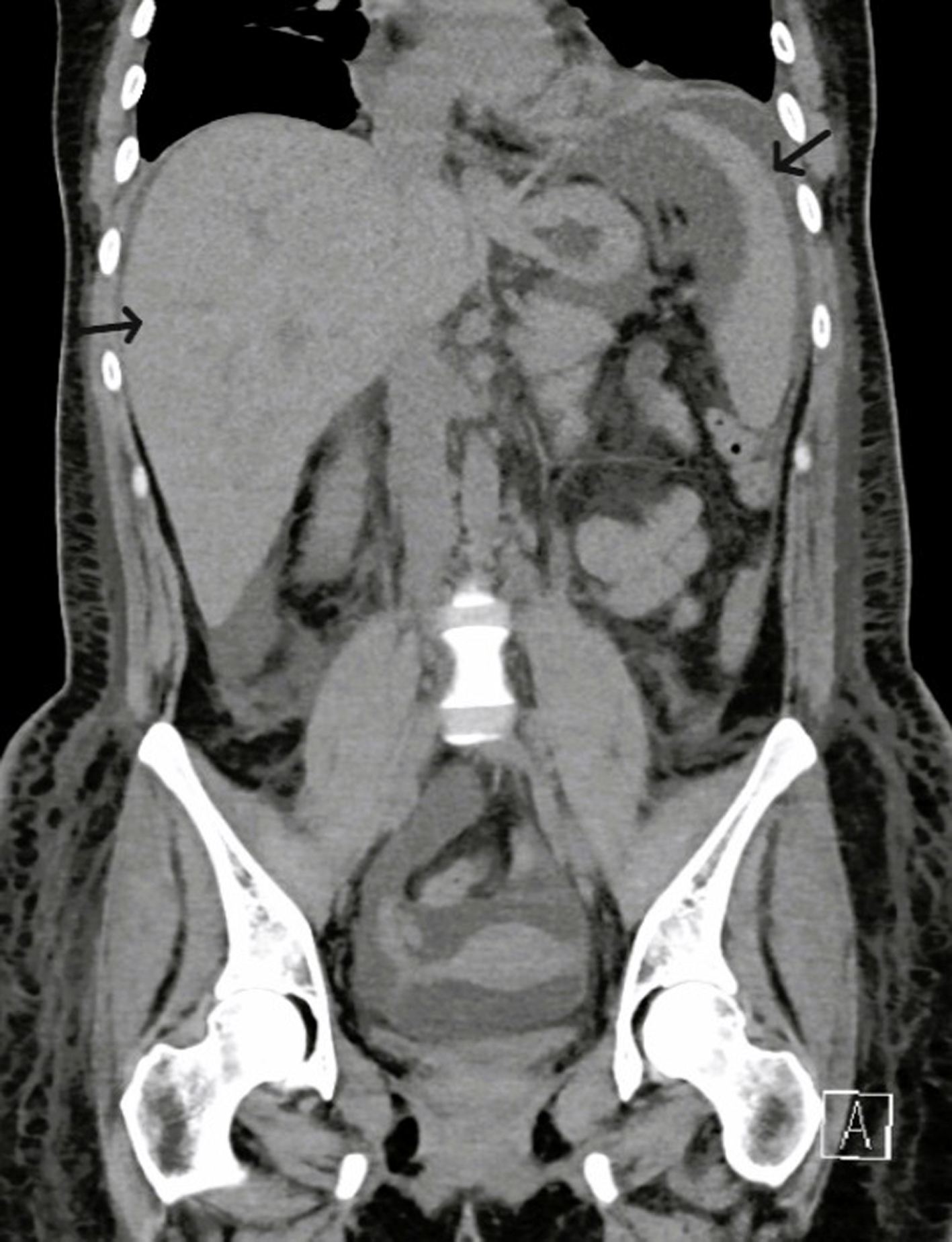
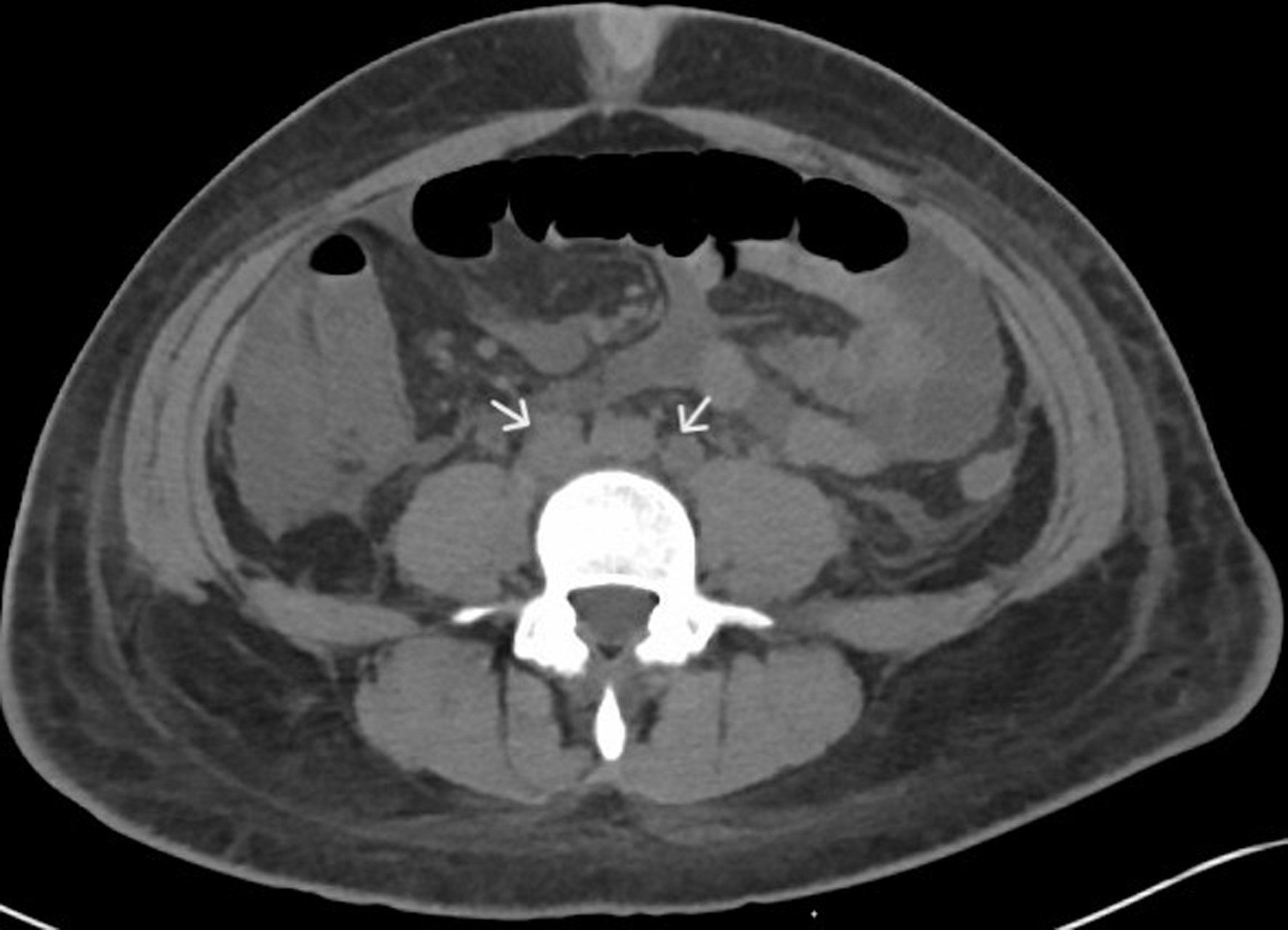
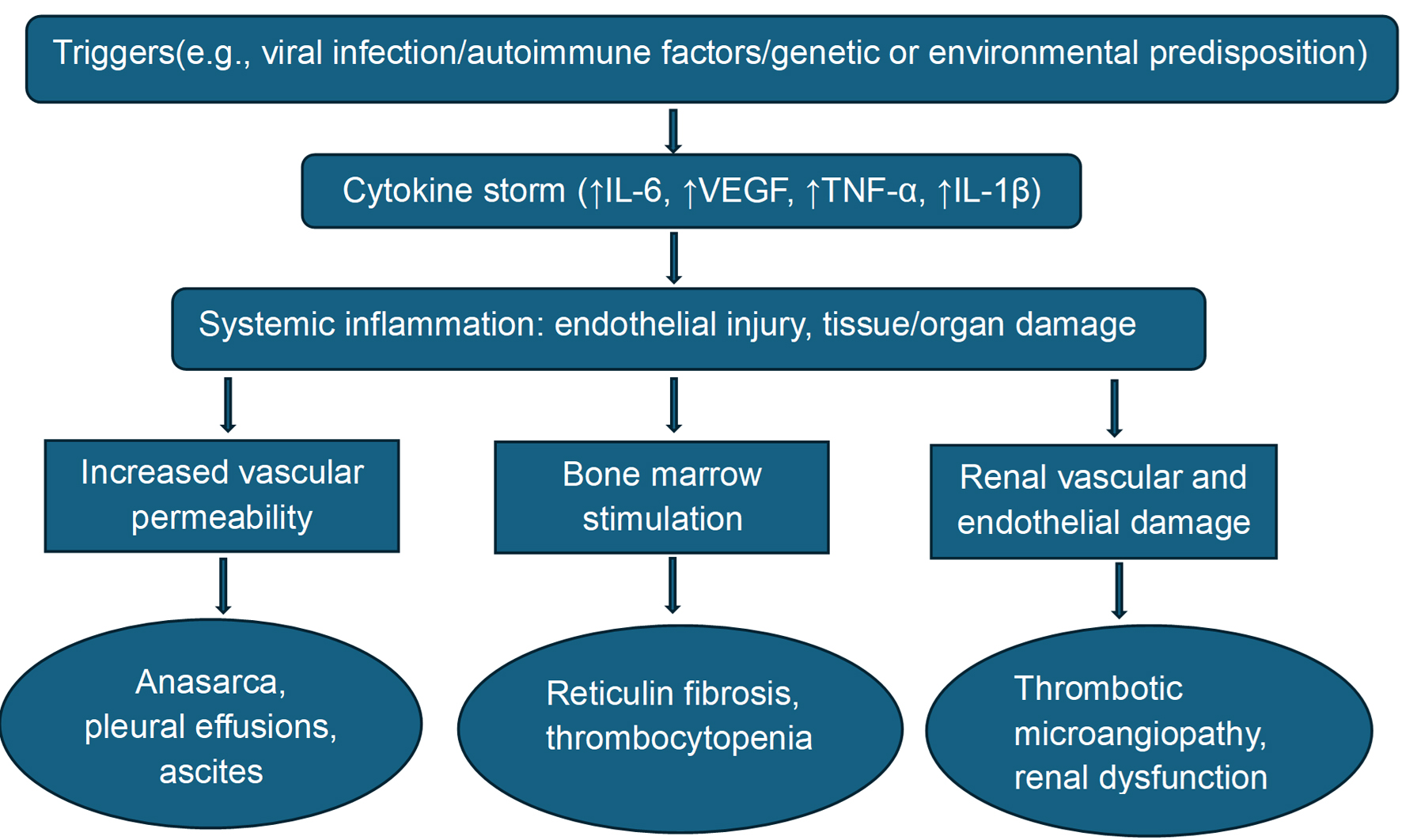
Figures

Tables
| Laboratory data | Admission | Days 7 - 10 | Discharge (day 33) | Normal range |
|---|---|---|---|---|
| CCP: cyclic citrullinated protein; SSA: Sjogren’s syndrome-related antigen A. | ||||
| White blood cells | 13.9 | 16.2 | 7.2 | 4.8 - 10.8 × 103 cells/µL |
| Hemoglobin | 9.8 | 6.8 | 8.4 | 13.5 - 17.5 g/dL |
| Platelet count | 144 | 50 | 113 | 130 - 400 × 103 cells/µL |
| Sodium | 128 | 129 | 139 | 136 - 145 mmol/L |
| Potassium | 4.6 | 3.6 | 4.1 | 3.5 - 5.1 mmol/L |
| Bicarbonate | 20 | 15 | 20 | 22 - 29 mEq/L |
| Blood urea nitrogen | 35 | 93 | 34 | 8 - 22 mg/dL |
| Creatinine | 1 | 3.46 | 0.59 | 0.7 - 1.2 mg/dL |
| Urine protein, random | 157 | 57.5 | - | mg/dL (no reference) |
| Urine protein-to-creatinine ratio | 0.87 | 1.24 | - | < 0.18 |
| Hemoglobin A1c | 5.5 | - | - | 4.4-5.6% |
| Total bilirubin | 0.6 | 0.6 | 0.4 | 0.20 - 1.00 mg/dL |
| Aspartate transaminase | 17 | 27 | 21 | 10 - 34 U/L |
| Alanine transaminase | 8 | 10 | 70 | 10 - 44 U/L |
| Calcium | 7.7 | 7.6 | 8.6 | 8.5 - 10.2 mg/dL |
| Total protein | 6.2 | 5.1 | 5.6 | 6.1 - 8.2 g/dL |
| Albumin | 1.7 | 2.2 | 3.5 | 3.3 - 5.2 g/dL |
| C-reactive protein | 16.7 | 4.9 | - | < 0.5 mg/dL |
| Ferritin | 505 | 1,581 | 778 | 12 - 114 ng/mL |
| Erythrocyte sedimentation rate | 61 | 12 | 6 | 2 - 37 mm/h |
| Anti-SSA antibodies | - | > 8.0 | - | 0.0 - 0.9 antibody index |
| Rheumatoid factor | 77 | - | - | < 14 IU/mL |
| Anti-CCP antibodies | - | 230 | - | 0.0 - 2.9 U/mL |
| Lactate dehydrogenase | - | 237 | 217 | 12 - 225 U/L |
| Interleukin-1β | - | < 6.5 | - | < 6.5 pg/mL |
| Soluable IL-2 receptor | - | 3,477 | 1,689 | 175 - 858 pg/dL |
| Interleukin-6 | - | 3.7 | < 2 | ≤ 2.0 pg/mL |
| Interleukin-8 | - | 7.3 | < 3 | ≤ 3.0 pg/mL |
| Tumor necrosis factor | - | 2.2 | 1.7 | ≤ 7.2 pg/mL |
| Type | Lymph node involvement | Histopathological subtypes | Systemic symptoms | Key features/prognosis |
|---|---|---|---|---|
| CD: Castleman disease; HHV-8: human herpesvirus-8; HIV: human immunodeficiency virus; IL-6: interleukin-6; iMCD: idiopathic multicentric Castleman disease; IPL: idiopathic plasmacytic lymphadenopathy; MCD: multicentric Castleman disease; NOS: not otherwise specified; POEMS: polyneuropathy, organomegaly, endocrinopathy, monoclonal gammopathy, and skin changes; TAFRO: thrombocytopenia, anasarca, fever, reticulin fibrosis/renal failure, and organomegaly; UCD: unicentric Castleman disease. | ||||
| UCD | Single area (localized) | Hyaline vascular; plasmacytic; mixed | +/- (in few patients) | Mostly asymptomatic, may present with localized symptoms (e.g., chest or abdominal pain from mass effect). Excellent prognosis. |
| MCD | Multiple regions | Hyaline vascular; plasmacytic; mixed | Yes (constitutional symptoms) | Associated with IL-6-driven hypercytokinemia, organ dysfunction, hepatosplenomegaly, cytopenias. Further classified as below. |
| HHV-8-associated MCD | Multiple regions | Same as above | Yes | Caused by HHV-8, often in HIV+ or immunocompromised individuals. Poor prognosis without HIV control. |
| HHV-8-negative MCD (iMCD) | Multiple regions | Same as above | Yes | Etiology unknown, HIV and HHV-8 negative. Needs special pathological, clinical, and lab criteria for diagnosis. Variable prognosis, better with early treatment. Further subclassified below. |
| iMCD-POEMS | Multiple regions | Typically plasmacytic. Plasma cell neoplasm; paraneoplastic syndrome, monoclonal; often with lambda light chain restriction | Yes | Polyneuropathy, organomegaly, skin changes, endocrinopathy; often with edema and ascites. Osteosclerotic lesions on imaging. Treated as POEMS. Variable prognosis, depending on the associated genetic alteration and tumor. |
| iMCD-TAFRO | Multiple regions (often small) | Mixed or hypervascular | Yes, with unique lab and clinical features | Normal gamma globulins. Guarded prognosis without early treatment. |
| iMCD-IPL | Multiple regions | Plasmacytic or mixed | Yes | Features: thrombocytosis, hypergammaglobulinemia. Intermediate prognosis. |
| iMCD-NOS | Multiple regions | Variable | Yes | Does not fit POEMS, TAFRO, or IPL. Mixed clinical and lab features; etiology unknown. Variable to good prognosis. |
| Oligocentric CD | 2 - 3 adjacent lymph node areas | Not fully defined | None or mild | Proposed subtype; overlaps more with UCD clinically and therapeutically. |
| Criteria type | Criteria details | Explanation |
|---|---|---|
| CMV: cytomegalovirus; CRP: C-reactive protein; EBV: Epstein-Barr virus; ESR: erythrocyte sedimentation rate; HHV-8: human herpesvirus-8; HIV: human immunodeficiency virus; IL-6: interleukin-6; iMCD: idiopathic multicentric Castleman disease; LDH: lactate dehydrogenase; RA: rheumatoid arthritis; sIL-2R: soluble interleukin-2 receptor; SLE: systemic lupus erythematosus; TAFRO: thrombocytopenia, anasarca, fever, reticulin fibrosis/renal failure, and organomegaly; VEGF: vascular endothelial growth factor. | ||
| Major criteria: both must be met | 1) Histopathological lymph node features consistent with Castleman disease | Hyaline vascular, plasmacytic, or mixed subtype |
| 2) Multicentric lymphadenopathy (≥ 2 lymph node regions) | Confirmed via imaging or physical exam | |
| Minor criteria: at least two of the following (with ≥ 1 laboratory abnormality) | Laboratory abnormalities (≥ 1 required): | |
| 1) Elevated CRP or ESR | Inflammatory markers | |
| 2) Anemia | Normocytic or microcytic | |
| 3) Thrombocytopenia or thrombocytosis | Either can occur | |
| 4) Hypoalbuminemia | Often due to IL-6-mediated hepatic suppression | |
| 5) Renal dysfunction | Elevated creatinine or proteinuria | |
| Clinical features (optional): | ||
| 1) Constitutional symptoms (fever, weight loss, fatigue, night sweats) | Common systemic symptoms | |
| 2) Hepatosplenomegaly | Detected clinically or via imaging | |
| 3) Fluid accumulation (ascites, pleural effusion, edema) | Seen in TAFRO variant | |
| 4) Lymphocytic interstitial pneumonitis or other organ involvement | Based on imaging/biopsy | |
| Exclusion criteria | All must be excluded: | |
| 1) HHV-8 infection (especially in HIV+ patients) | By immunohistochemistry or PCR testing | |
| 2) Malignancies (e.g., lymphoma, metastatic cancer) | Ruled out by biopsy or imaging | |
| 3) Autoimmune/connective tissue diseases | E.g., SLE, Sjogren’s, RA assessed via serologies and clinical history | |
| 4) Infectious diseases (e.g., EBV, CMV, tuberculosis, sepsis) | Should not explain presenting findings | |
| Additional laboratory abnormalities that support the diagnosis but are not required | 1) Elevated IL-6, sIL-2R, VEGF, IgA, IgE, LDH, and/or beta-2 microglobulin | |
| 2) Reticulin fibrosis on bone marrow biopsy (particularly in patients with TAFRO syndrome) | ||
| Type | Description | Key features |
|---|---|---|
| ANA: antinuclear antibody; CMV: cytomegalovirus; CRP: C-reactive protein; EBV: Epstein-Barr virus; HHV-8: human herpesvirus-8; IL-6: interleukin-6; iMCD: idiopathic multicentric Castleman disease; MDS: myelodysplastic syndrome; SLE: systemic lupus erythematosus; SSA: Sjogren’s syndrome-related antigen A; TAFRO: thrombocytopenia, anasarca, fever, reticulin fibrosis/renal failure, and organomegaly. | ||
| TAFRO with definite iMCD (iMCD-TAFRO) | Meets TAFRO clinical criteria with confirmed iMCD on lymph node biopsy (typically hypervascular type). | Histologically proven; IL-6 elevation; requires full workup including biopsy. Lymph nodes are usually smaller compared to other types of iMCD. |
| TAFRO with possible iMCD | TAFRO clinical features present but lymph node biopsy unavailable or non-diagnostic. | Clinical suspicion is high; supportive labs (e.g., IL-6, CRP); often treated as iMCD-TAFRO. |
| TAFRO associated with autoimmune diseases | Coexists with autoimmune diseases (e.g., Sjogren’s, SLE); may mimic or overlap with iMCD. | Positive autoantibodies (ANA, SSA, etc.); may respond to immunosuppression. |
| TAFRO associated with infections | Associated with infections (e.g., EBV, CMV, HHV-8); clinical mimic of TAFRO syndrome. | Viral serologies positive; often younger patients; may require antiviral or supportive care. |
| TAFRO-like syndrome | Mimics TAFRO features but occurs in malignancies or other systemic diseases (e.g., lymphoma, MDS). | Non-idiopathic; underlying pathology explains presentation; prognosis varies widely. |
| Manifestation | Description | Workup |
|---|---|---|
| CRP: C-reactive protein; CT: computed tomography; ESR: erythrocyte sedimentation rate; MPGN: membranoproliferative glomerulonephritis; TAFRO: thrombocytopenia, anasarca, fever, reticulin fibrosis/renal failure, and organomegaly; TMA: thrombotic microangiopathy. | ||
| Thrombocytopenia | Ranges from moderate to severe; may lead to bleeding. | Complete blood count, peripheral blood smear |
| Anasarca | Generalized edema, pleural effusions, and ascites. Thoracocentesis and paracentesis are often required. | Physical examination, chest X-ray, CT chest, abdomen and pelvis |
| Fever and systemic inflammation | High-grade fevers with elevated CRP, ESR, and ferritin. | Physical examination, laboratory analysis |
| Reticulin fibrosis and marrow changes | Pathology shows fibrosis and megakaryocytic hyperplasia, often with larger cells. | Bone marrow biopsy |
| Renal dysfunction | Acute kidney injury. TMA and MPGN on biopsy. | Laboratory analysis, urine studies, renal biopsy |
| Organomegaly | Mild to moderate hepatosplenomegaly, and lymphadenopathy (could be multifocal). | Physical examination, laboratory analysis, CT chest, abdomen, and pelvis |
| Other symptoms and findings | Fatigue, weight loss, nausea, vomiting, anemia, anorexia, and hypoalbuminemia. | Physical examination, laboratory analysis |
| Autoimmune overlap | Patients may meet criteria for associated autoimmune conditions, such as Sjogren’s syndrome, or have features of their acute flare. | Physical examination, laboratory analysis |
| Category | Criteria | Details |
|---|---|---|
| CRP: C-reactive protein; iMCD: idiopathic multicentric Castleman disease; MCD: multicentric Castleman disease; TAFRO: thrombocytopenia, anasarca, fever, reticulin fibrosis/renal failure, and organomegaly; TMA: thrombotic microangiopathy. | ||
| Major criteria | All three must be present: | |
| Thrombocytopenia | Platelet count ≤ 100,000/µL | Persistent or progressive. |
| Anasarca | Ascites, pleural effusion, and/or generalized edema | Clinically and/or radiologically detected. |
| Systemic inflammation | Fever ≥ 37.5 °C and/or CRP ≥ 2.0 mg/dL | Evidence of systemic inflammatory response. |
| Minor criteria (at least two required) | At least two must pe present: | |
| Reticulin fibrosis | Bone marrow biopsy findings | Bone marrow biopsy shows reticulin myelofibrosis or megakaryocytic hyperplasia. |
| Renal dysfunction | Elevated serum creatinine (> 1.5 mg/dL) or proteinuria | Often reflects TMA on biopsy. |
| Mild organomegaly | Mild hepatomegaly, splenomegaly, and/or lymphadenopathy | Less prominent than in classic MCD or other types of iMCD. |
| Differential diagnosis | Key features that overlap | Distinguishing features |
|---|---|---|
| aHUS: atypical hemolytic uremic syndrome; ANA: antinuclear antibody; C3, 4: complement 3, 4; ds-DNA: double-stranded deoxyribonucleic acid; HLH: hemophagocytic lymphohistiocytosis; iMCD: idiopathic multicentric Castleman disease; LDH: lactate dehydrogenase; MAS: macrophage activation syndrome; POEMS: polyneuropathy, organomegaly, endocrinopathy, monoclonal gammopathy, and skin changes; SLE: systemic lupus erythematosus; TAFRO: thrombocytopenia, anasarca, fever, reticulin fibrosis/renal failure, and organomegaly; TMA: thrombotic microangiopathy; VEGF: vascular endothelial growth factor. | ||
| Classic iMCD | Lymphadenopathy, systemic inflammation | Indolent course, more prominent lymph node involvement |
| SLE | Cytopenias, serositis, renal dysfunction | ANA/anti-dsDNA positivity, SLE-specific criteria: anti-Smith antibody, typical malar rash, low C3/C4, positive antiphospholipid antibodies |
| POEMS syndrome | Organomegaly, edema, polyneuropathy | Monoclonal plasma cell disorder, VEGF levels, and sclerotic bone lesions |
| Lymphoma | Lymphadenopathy, constitutional symptoms | Clonal lymphoid cells on biopsy, B symptoms |
| Severe infection (e.g., sepsis) | Fever, multiorgan dysfunction | Positive cultures, response to antimicrobials |
| aHUS | Thrombocytopenia, anemia, systemic inflammation, elevated LDH, TMA, renal failure (more acute in aHUS) | Dysregulation of the complement pathway: often low C3, normal or mildly low C4, absent or minimal lymphadenopathy, organomegaly is uncommon, and anasarca is rare. |
| HLH/MAS | Cytopenias, fever, hyperferritinemia | Extremely high ferritin, low natural killer cell activity, HLH criteria |
| Treatment modality | Indication | Complications |
|---|---|---|
| CHOP: cyclophosphamide, doxorubicin, vincristine, prednisone; IL-6: interleukin-6; JAK: Janus kinase; TAFRO: thrombocytopenia, anasarca, fever, reticulin fibrosis/renal failure, and organomegaly. | ||
| Glucocorticoids [7, 15, 20-22] | First-line for acute flare | Hyperglycemia, infections, and hypertension |
| Anti-IL-6 (siltuximab/sarilumab/tocilizumab) [24-29] | IL-6 blockade in moderate-severe disease | Risk of infection, neutropenia |
| Anti-IL-1 (anakinra/canakinumab) [38-40] | Refractory or exaggerated cytokine storm, autoimmune overlap, IL-6 inhibitor failure | Infections, injection site reactions |
| Cyclosporine [30-32] | Often used in combination with steroids; steroid/anti-IL-6 refractory cases | Nephrotoxicity, hypertension |
| Rituximab [33-37] | Autoimmune overlap or B-cell involvement. Used alone or in combination with siltuximab and steroids. | Infusion reactions, immunosuppression |
| Cyclophosphamide [41, 42] | Severe or refractory disease. Part of CHOP or in combination with bortezomib and steroids | Myelosuppression, infertility |
| CHOP regimen [43-45] | Suspected or confirmed overlap with lymphoma or severe inflammation | Toxicity, immunosuppression |
| JAK inhibitors (ruxolitinib) [25, 27] | Experimental, used in cases refractory to anti-IL-6 therapy. Successful in sporadic cases | Cytopenias, thromboembolism |
| Treatment modality | Summary of patient response | Type of study |
|---|---|---|
| IL-6: interleukin-6; TAFRO: thrombocytopenia, anasarca, fever, reticulin fibrosis/renal failure, and organomegaly. | ||
| Tocilizumab (IL-6 inhibitor) | In a systematic review of 31 patients, approximately 51.6% achieved complete response, while others had partial or no response, and some succumbed to the disease [28]. In another case with severe liver dysfunction, significant clinical improvement was noted with tocilizumab, suggesting its potential utility in TAFRO syndrome [29] | Systematic review |
| Siltuximab (IL-6 inhibitor) | In a randomized trial, results showed that siltuximab combined with supportive care was more effective and well tolerated than supportive care alone in symptomatic multicentric Castleman disease, offering a valuable new treatment option, including TAFRO syndrome [23]. Positive response noted at 3 months in a patient with severe renal involvement [26]. | Clinical trial, case report |
| Anakinra (IL-1 inhibitor) | Two pediatric patients with severe, rapidly progressing TAFRO syndrome
achieved remission with high-dose intravenous anakinra [40]. Another adult patient with life-threatening TAFRO syndrome showed rapid clinical improvement following anakinra therapy [39]. |
Case reports |
| Cyclosporine | Three cases that were refractory to anti-IL-6 therapy, treated with corticosteroids and cyclosporine, experienced resolution of anasarca and improved renal function [30-32]. | Case reports |
| Rituximab | In one case, a patient responded well to rituximab combined with
prednisolone as first-line treatment [33]. Multiple other case reports reported use in relapsed and refractory scenarios [34-37]. |
Case reports |
| Combination therapy | Combination of tocilizumab and siltuximab was successfully used in a
patient with severe disease, without the need for immunomodulators or aggressive supportive care
[24]. A severe case of TAFRO syndrome was successfully treated with a combination of rituximab, steroid pulse therapy, plasma exchange, and romiplostim, with rapid improvement of renal function, anasarca, and thrombocytopenia. In a case of severe-grade TAFRO syndrome, administration of triple therapy with corticosteroid, tocilizumab, and cyclosporine achieved successful remission of the disease with discontinuation of hemodialysis by week 5 and improved platelets by week 9 [46]. |
Case reports |